About the CMAP File Format
SimpleSynteny uses BLAST to map your query genes/proteins onto your genome(s) and stores these coordinates in a human-readable format called a 'CMAP' (Contig-Mapping) file. You can also use 'Advanced Mode' to manually write your own CMAP files and generate a more customized image. Each CMAP file represents a single genome, drawn as a single row of one or more contigs in a figure. The program processes each CMAP in order, drawing genomes in the image from top-to-bottom. Here's how the file format works:
Mapping a Single Contig: Each line of a CMAP file represents a single contig/supercontig which is drawn from left-to-right. The format of a basic contig line starts with the name of the contig, followed by a space, and then data contained within { } curly-braces as follows: MyContigName {FirstContigBasePair# ... LastContigBasePair#}. The numbers just inside the { } curly-braces represent the first (typically 1) and last base pair (BP) numbers of the contig, respectively. Between these numbers reside one or more query genes or proteins to map onto the contig.Here's an example figure followed by the contig line used to make it:

G4 {1 [ 412|412 (1|1 <== T1 98|98) 509|509 ] [ 801|801 (1|1 T2 ==> 35|59) 860|882 ] 882}
We can see the contig (shown in light beige) is named in the top-left corner as 'G4' and contains two query protein sequences (DNA sequences could also have been used). The CMAP line denotes the contig stretches from BP's 1-882, however, to optimize space the program removed long stretches of the contig in the figure (BP's 1-398 and 525-785) since they did not contain any query sequence information. Anytime the program removes a section of a contig to save space, the contig is drawn with a jagged edge and corresponding BP numbers are indicated beneath the contig. If the end of a contig is reached, a straight edge is shown as seen above BP 882. The query protein 'T1' is shown in red and faces upstream, while protein 'T2' is shown in blue and faces downstream. Every query sequence marked on a contig line is placed in square brackets [ ] and separated by two spaces. For a given query, the information inside the brackets is used to decide where on the contig to draw a box to represent it, if and where it should be shaded, what direction it faces (5'-to-3' vs 3'-to-5'), and what it is named. Using 'T2' from the example above, let's work our way from the query name outwards to see what everything means.
Query Names and Direction: Query names are automatically capitalized and italicized by the program for figures even if they are spelled in lowercase on a CMAP line. The spelling of names is important, as the program looks for matching identical names between genomes and, if it finds them, draws a connecting line between them. Currently, it is NOT recommend to have the same name occur more than once in the same genome/CMAP file (this could easily happen with a common gene like aldolase for example) as images quickly get very confusing. To prevent this, if you have the same gene name repeated more than once, SimpleSynteny will try to append a ".1, .2, .3" etc. to each new occurrence and treat them as separate genes. To the left or right of the gene name will respectively be a <== or ==> direction arrow (separated from the name by one space). These are used to draw the gene direction as found by BLAST or you can set it manually when using Advanced Mode. Two spaces are used to separate the gene name and direction arrow from any numbers.
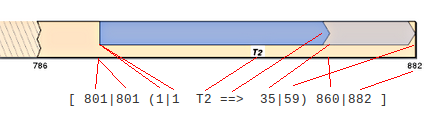
Contig Numbering: Continuing outside the ( ) parenthesis, a single space separates similar sets of numbers used to indicate where on the contig to draw the box. These are simply the contig BP numbers that correspond to the start and stop of the shading and full-length query numbers as discussed above. Again, the numbers closest to the query name refer to shading, while those on the other side of the | symbol correspond to the full-length query sequence. When using DNA query sequences, the difference between the first and last shading and full-length numbers should be the same for the contig space as it is for the query space. Since our example query sequences are proteins, the difference between the first and last contig DNA coordinates have been multiplied by three to account for codons. It is important to note that SimpleSynteny's regular mode assumes there are no introns in coding sequences. If you need to take introns into consideration you will need to manually edit your CMAPs using Advanced Mode and represent each exon as a separate entry. One more space in each direction is used to separate the final [ ] square brackets which denote the boundary of the query entry.
We can now consider this query like the following line:
Multiple genes can be inserted inside a contig, they just need to be separated by two spaces on either side of them.
Valid Numbers: In order for a CMAP line to be valid, numbers to the left of the query name should be less than the corresponding numbers on the right. For example, the first contig BP number must always be less than the end BP number. Query sequences must not be longer than the size of their residing contig, and shading numbers must always be equal to or fit within the range of the full-length sequence.
Removing Gene Directions: Advanced Mode also allows you to remove directionality from a gene entirely by placing == on both sides of a gene name. The following example shows Ami1 drawn as a rectangle without direction:

Contig1 {1 [ 2193|3193 (1|1 == Ami1 == 2351|6351) 7685|8685 ] [ 12393|12393 (1|1 Cia30 ==> 756|756) 13148|13148 ] 14000}
Now that you understand the CMAP format, you can use Advanced Mode to make the following adjustments to your image:
- Edit or remove unwanted query fragments found by BLAST with the main program (use the CMAPs provided with the original output as a starting template).
- Add in separate entries to represent introns and exons.
- Customize gene shading to highlight particular areas of interest.
- Remove gene directions and display them as basic rectangles.
- Manually insert genes of importance if you need to use a certain BLAST E-value cutoff but a gene of interest is being left out.
- Make manual images for teaching purposes etc.
If you encounter any problems with SimpleSyntey please e-mail Dan Veltri at: Dan.Veltri@gmail.com. Please include "SimpleSynteny" in the subject line and include your CMAP files in addition to a detailed explanation of the problem so we can better help you. We also encourage feedback for changes and/or improvements to the program.